Hereditary Spastic Paraplegia
NEURO · EP 06 · NEUROLOGY
Before You Listen
- Prerequisites: upper vs lower motor neuron sign patterns; corticospinal tract anatomy and dorsal column organization (long-tract neuroanatomy from NEURO-01); MS, ALS, GBS, CIDP, myasthenia, Lambert-Eaton, and FND distinctions from NEURO-01 through NEURO-05.
- Runtime: 48 minutes 56 seconds.
- Topic in one line: the dying-back axonopathy model of hereditary spastic paraplegia (HSP), the pure vs complicated Harding classification, the four board-tested spastic paraplegia gene (SPG) loci (SPG4, SPG3A, SPG7, SPG11), the M-DASH-H differential, the rehabilitation pillars of HSP, and a focused look at the spinocerebellar ataxias as the cerebellar-dominant look-alike.
Vignette. A 34-year-old man presents with a 7-year history of slowly progressive lower-extremity stiffness, urinary urgency, and difficulty running. His father required a cane by age 50. Examination shows symmetric LE spasticity with bilateral hyperreflexia, sustained ankle clonus, bilateral Babinski signs, mildly impaired vibration sense at the great toes, scissoring gait with toe-walking, and completely normal upper extremities, normal cognition, normal bulbar function, and normal optic discs. Brain and full-spine MRI show only thoracic cord atrophy without focal lesions. EMG/NCS are normal. B12, methylmalonic acid, very long-chain fatty acids (VLCFAs), and HTLV-1 serology are negative.
What is the most likely diagnosis, the most likely gene, and the single feature that most strongly excludes MS?
Section 1 — The Dying-Back Axonopathy and Pure vs Complicated HSP
Bottom line: the longest CNS axons (corticospinal tract to LE, dorsal column gracilis) starve first when intracellular logistics fail, explaining bilateral LE spasticity, distal vibration loss, and urinary urgency in pure HSP.
Hereditary spastic paraplegia (also called familial spastic paraplegia or Strumpell-Lorrain disease) is a clinically and genetically heterogeneous group of inherited neurodegenerative disorders unified by progressive spasticity and weakness of the lower extremities caused by length-dependent axonal degeneration of the corticospinal tracts and, variably, the dorsal columns. Prevalence is roughly 1.2 to 9.6 per 100,000. More than 80 genetic loci (SPG1 through SPG80+) have been identified, yet the pure phenotype is remarkably consistent across them.
The unifying mechanism is the dying-back axonopathy model. The corticospinal neurons projecting from motor cortex to lumbosacral cord segments have axons that may exceed one meter in length. These axons depend on continuous intracellular logistics: mitochondrial delivery for energy, microtubule-based axonal transport, endoplasmic reticulum membrane shaping, and autophagic clearance. When any of these processes fails, the distal portions farthest from the cell body starve first. Lower-extremity tracts degenerate before and more severely than upper-extremity tracts; distal segments fail before proximal. Postmortem confirms maximal corticospinal degeneration at thoracic and lumbar levels with relative preservation at cervical levels, and degeneration of the fasciculus gracilis (LE proprioception/vibration) with sparing of the fasciculus cuneatus (UE).
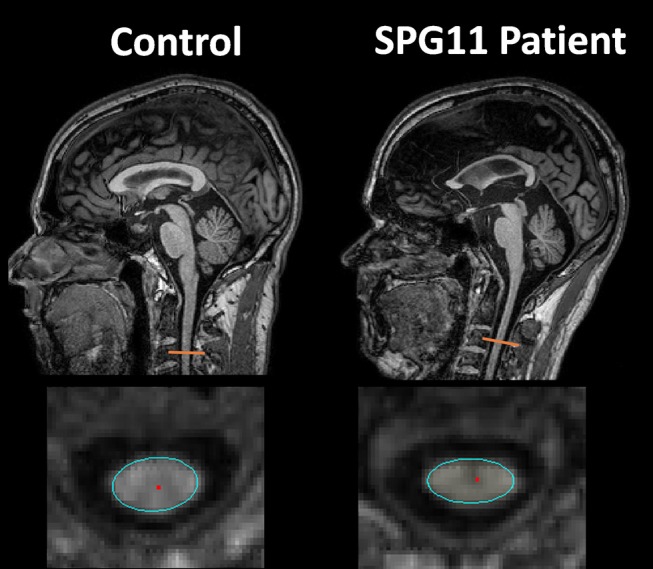
Source: da Graça FF, de Rezende TJR, Vasconcellos LFR, Pedroso JL, Barsottini OGP, França MC Jr. “Neuroimaging in Hereditary Spastic Paraplegias: Current Use and Future Perspectives.” Frontiers in Neurology. 2019;9:1117. Figure 1. doi:10.3389/fneur.2018.01117. Licensed under CC BY 4.0. https://pmc.ncbi.nlm.nih.gov/articles/PMC6346681/
| Cellular pathway | Representative SPG genes | Protein | Failure mode |
|---|---|---|---|
| ER membrane shaping | SPG4, SPG3A, SPG31, SPG12 | Spastin, atlastin-1, REEP1, reticulon-2 | Disrupted tubular ER along the axon |
| Microtubule severing / axonal transport | SPG4, SPG10 | Spastin, kinesin heavy chain | Failed delivery of cargo to the distal axon |
| Mitochondrial quality control | SPG7 | Paraplegin | Energy failure, oxidative stress |
| Autophagy / lysosomal reformation | SPG11, SPG15 | Spatacsin, spastizin | Accumulation of autophagic debris |
| Lipid metabolism | Multiple | Cholesterol/phospholipid enzymes | Membrane synthesis failure |
The Harding classification splits HSP into pure and complicated forms, and this distinction drives the differential, the genetic testing strategy, and the prognosis. Pure HSP accounts for 70-80% of cases and is defined by a tight clinical triad with everything else explicitly normal.
| Feature | Pure HSP | Complicated HSP |
|---|---|---|
| LE spasticity + weakness | Yes (bilateral, symmetric) | Yes |
| Mild dorsal column loss (vibration > proprioception, distal LE) | Yes | Yes |
| Urinary urgency / frequency (60-80%) | Yes (neurogenic detrusor overactivity) | Yes |
| Cognition | Normal | May be impaired (SPG11, SPG15, SPG21) |
| Upper extremities | Normal | May be involved |
| Bulbar function | Normal | May be involved |
| Cerebellar / ataxia | Normal | May be present (SPG7, SPG15) |
| Optic atrophy | Absent | May be present (SPG7) |
| Peripheral neuropathy | Absent | May be present (SPG7, SPG10, SPG11) |
| Thin corpus callosum on MRI | Absent | Hallmark of SPG11 |
| Onset | Adult more common | Earlier onset typical |
| Progression | Slow, decades | Faster, more guarded |
| Inheritance | Often autosomal dominant | Often autosomal recessive |
The features that are absent in pure HSP are as diagnostically important as those that are present. A patient with progressive spastic paraparesis plus bulbar signs is not pure HSP. Think ALS. UE involvement points to MS or cervical myelopathy. Cognitive decline points to complicated HSP (especially SPG11 if a thin corpus callosum is present). The pure form is a strictly length-dependent disease confined to the longest tracts; anything outside that pattern reclassifies the case.

Clinical Pearl — Why the bladder gets caught up in a “leg disease”

The corticospinal tract carries not only voluntary motor commands to the legs but also the inhibitory descending signals to the detrusor. When these descending brakes are lost, the detrusor (the muscle of the bladder wall) becomes hyperactive: neurogenic detrusor overactivity, present in up to 60-80% of pure HSP patients, often preceding or accompanying motor symptoms.
Board Trap — “Spastic paraparesis with bulbar signs = HSP”
False. Pure HSP is strictly length-dependent with normal bulbar function, normal UE, and normal cognition. Bulbar involvement should redirect the differential to ALS; UE involvement to MS or cervical cord pathology; cognitive decline to complicated HSP (SPG11 if thin corpus callosum on MRI).
High Yield — Pure HSP triad and exclusions

- Triad: bilateral symmetric LE spasticity + mild distal vibration loss > proprioception loss + urinary urgency (60-80%).
- All normal: cognition, UE, bulbar function, cerebellar function, vision, no LMN signs, no sensory level.
- Pathology: length-dependent dying-back of corticospinal tract and fasciculus gracilis.
- 70-80% of HSP is the pure form; 20-30% is complicated.
But what is equally important is what you do not see on the MRI. Exactly, you are looking for negative findings. There should be no focal lesions, there should be no T2-hyperintense plaques, and there must be absolutely no contrast enhancement when you give gadolinium.
— NEURO-06 podcast, ~25:29