PEDS · EP 04 · NMD
Before You Listen
Episode Setup
- Topic in one line: Part 2 of the pediatric neuromuscular disease (NMD) tour continues distal to the anterior horn and works outward through the motor unit. The peripheral nerve section covers Charcot-Marie-Tooth (CMT) disease and Friedreich ataxia (FRDA), including the 38 m/s nerve conduction velocity cutoff that separates demyelinating CMT1 from axonal CMT2, the PMP22 gene-dosage story behind CMT1A and its mirror-image deletion in hereditary neuropathy with liability to pressure palsies (HNPP), and the FRDA pentad of areflexia plus Babinski plus hypertrophic cardiomyopathy plus scoliosis plus diabetes. The muscle-disorder section then turns to myotonic dystrophy type 1 (DM1), the congenital myopathies (central core, nemaline, myotubular, Schwartz-Jampel), and Pompe disease, anchored by the new American College of Medical Genetics (ACMG) 2024 CTG repeat thresholds and the ryanodine receptor 1 (RYR1) link between central core disease and malignant hyperthermia. The chapter closes with the anesthesia and exercise-prescription principles that unify everything covered in Part 1 and Part 2: succinylcholine is absolutely contraindicated in the dystrophinopathies and the RYR1 myopathies, total intravenous anesthesia (TIVA) with rocuronium is the safe combination, submaximal aerobic exercise (swimming, flat cycling) is encouraged, and eccentric and high-resistance work must be avoided to prevent overwork weakness.
- Prerequisites: Part 1 of PEDS-04 (dystrophinopathies and the spinal muscular atrophy (SMA) classification), the motor unit anatomy from EDX-01, the difference between myopathic and neurogenic electromyography (EMG) patterns, and the basic genetics of autosomal dominant, autosomal recessive, and X-linked inheritance plus trinucleotide repeat expansion biology.
- Runtime: 1 hour 14 minutes (combined runtime across Part 1 and Part 2).
Vignette. A 9-year-old girl is referred for “clumsy walking” and recurrent ankle sprains. Her parents note that she trips frequently and wears out the lateral edges of her shoes. On examination she has bilateral pes cavus with hammertoes, an “inverted champagne bottle” silhouette to her lower legs with prominent peroneal atrophy, foot drop with a steppage gait, depressed deep tendon reflexes throughout, and reduced vibratory sensation to the mid-shin bilaterally. Strength is 5/5 proximally, 3/5 at the ankle dorsiflexors. Her father had the same foot shape and used to wear braces. Nerve conduction studies show a median motor conduction velocity of 22 m/s with reduced amplitude, and electromyography shows chronic neurogenic changes distally. A muscle biopsy is not performed.
Name the most likely diagnosis and its specific subtype, state the underlying genetic mechanism and the chromosomal location, predict what a nerve biopsy would have shown if performed, identify the single chemotherapeutic agent that is absolutely contraindicated in this disease, and outline the rehabilitation cornerstone.
(Answer at the end of this chapter)
Section 3: Charcot-Marie-Tooth Disease and Hereditary Neuropathies
Bottom line: Charcot-Marie-Tooth (CMT) disease is the most common inherited peripheral neuropathy at approximately 1 in 2,500 with the unifying syndrome of length-dependent distal motor and sensory neuropathy producing pes cavus, hammertoes, peroneal atrophy (“inverted champagne bottle” legs), distal weakness with foot drop, and stocking-glove sensory loss; the fundamental classification splits demyelinating CMT (median motor nerve conduction velocity below 38 m/s) from axonal CMT (above 38 m/s); CMT1A is the most common subtype (approximately 50 percent) caused by autosomal dominant peripheral myelin protein 22 (PMP22) duplication at 17p11.2 with onion bulb formation on biopsy; CMT2A is the most common axonal subtype (MFN2); vincristine is absolutely contraindicated in CMT because it accelerates the neuropathy; hereditary neuropathy with liability to pressure palsies (HNPP) is the reciprocal deletion disorder with tomaculous nerve biopsy.
The unifying CMT syndrome. Hereditary motor and sensory neuropathy (HMSN), now universally called Charcot-Marie-Tooth disease, has a population prevalence of approximately 1 in 2,500, making it the most common inherited disorder of peripheral nerve. More than one hundred causative genes have been identified, but the clinical syndrome is conserved across subtypes: a slowly progressive, length-dependent, distal-predominant motor and sensory neuropathy that hits the longest axons first and works centrally over years. The classic presenting child has pes cavus (a high medial longitudinal arch), hammertoes (hyperextension at the metatarsophalangeal joint with flexion at the proximal interphalangeal joint), and bilateral peroneal atrophy producing the “inverted champagne bottle” leg silhouette, in which the proximal thigh stays well-developed while the distal calf wastes to bone and tendon. Gait shows a steppage pattern from foot drop, with the child lifting the knee high to clear a plantarflexed foot. Reflexes are depressed or absent first at the ankle, then progressively higher. Sensory loss is stocking-distribution and includes vibration, joint position, and pinprick. Hand involvement follows a similar pattern, with intrinsic atrophy and clumsy fine-motor control emerging later in adolescence and adulthood.
The 38 m/s cutoff. The single most important board fact in CMT classification is electrophysiologic. The median motor nerve conduction velocity (NCV) threshold of 38 m/s divides the entire disorder into demyelinating and axonal forms. Below 38 m/s, the slowing reflects loss of myelin insulation, and the diagnosis is CMT1 (autosomal dominant demyelinating), CMT3 (severe demyelinating, formerly Dejerine-Sottas), or CMT4 (autosomal recessive demyelinating). Above 38 m/s, conduction velocities are preserved or only mildly slowed because the surviving axons are myelinated normally; the compound muscle action potential amplitude is reduced because there are simply fewer axons. That is CMT2, the axonal form. CMTX1 sits in an intermediate range (25 to 40 m/s in affected males) and is X-linked, often the right answer when a family pedigree shows skipped generations and severely affected males with milder female carriers.
| Subtype | Pathophysiology | Inheritance | NCV (median motor) | Gene | Histology |
|---|---|---|---|---|---|
| CMT1A | Demyelinating | AD | <38 m/s (often 15-30) | PMP22 duplication, 17p11.2 | Onion bulbs |
| CMT1B | Demyelinating | AD | <38 m/s | MPZ, 1q22 | Onion bulbs |
| CMT2A | Axonal | AD | >38 m/s | MFN2 (mitofusin 2), 1p36 | Axonal degeneration |
| CMT3 (Dejerine-Sottas) | Severe demyelinating | AD or AR | <10 m/s | PMP22, MPZ, EGR2 | Prominent onion bulbs |
| CMT4 | Demyelinating | AR | <38 m/s | Multiple | Onion bulbs |
| CMTX1 | Intermediate | X-linked | 25-40 m/s (males) | GJB1 (connexin 32), Xq13 | Mixed |
CMT1A and the PMP22 gene-dosage story. CMT1A accounts for roughly half of all CMT cases. The mechanism is unusual and worth understanding because it explains a separate disease in the same chromosomal neighborhood. A 1.5-megabase duplication on chromosome 17p11.2 creates three functional copies of the peripheral myelin protein 22 (PMP22) gene rather than the normal two. The extra dose of PMP22 disrupts the stoichiometry of compact myelin, destabilizes the insulation, and produces the chronic demyelination-remyelination cycles that show up histologically as onion bulbs — concentric layers of Schwann cell processes wrapped around an axon like the rings of a sliced onion, a visible record of repeated myelin repair. The reciprocal lesion at the same locus, a 1.5-megabase deletion, produces hereditary neuropathy with liability to pressure palsies (HNPP): only one functional copy of PMP22, dose-insufficient myelin, and an episodic phenotype rather than a progressive one. HNPP patients develop transient compressive mononeuropathies at vulnerable sites (carpal tunnel, peroneal palsy at the fibular head, ulnar palsy at the elbow) after trivial mechanical stress, often recovering over weeks. The biopsy hallmark of HNPP is the tomaculum (Latin for sausage), a focal, sausage-shaped swelling of redundant myelin folds along the internode.
So they have an extra copy of the PMP22 gene, and that overproduction somehow destabilizes the myelin insulation. And because the body is constantly trying to repair this breaking myelin, a nerve biopsy will show classic onion bulb formations. The Schwann cells wrap layer after layer of scar tissue around the nerve, looking exactly like the rings of a sliced onion under the microscope.
— PEDS-04-b podcast, ~41:17
CMT2A. The most common axonal subtype is caused by mutations in the MFN2 (mitofusin 2) gene at 1p36, encoding a mitochondrial outer membrane fusion protein. The axon depends on dense, healthy mitochondrial trafficking to keep its longest segments alive, so a fusion defect produces selective length-dependent axonal degeneration. Phenotypically, CMT2A looks much like CMT1A from across the room (pes cavus, foot drop, steppage gait, depressed reflexes), but the NCV pattern is different. Conduction velocities are normal or only mildly slowed because the surviving axons are myelinated normally; the compound muscle action potential amplitudes are reduced in proportion to axonal loss. Several other autosomal dominant and recessive genes contribute to the CMT2 family, but MFN2 is the canonical board answer.
The vincristine rule. Every resident should know one pharmacogenomic fact about CMT cold. Vincristine is absolutely contraindicated in any patient with CMT. Vincristine binds tubulin and disrupts axonal microtubule transport, and in a peripheral nerve that is already operating with reduced reserve, even a single standard dose can convert a walking patient with foot drop into a wheelchair-dependent patient with global flaccid weakness within days. The damage is often irreversible. Every patient with a CMT diagnosis should carry medical alert identification, and every oncology consultation should screen for inherited neuropathy when vincristine is on the table. Other agents that carry less catastrophic but still real risk in CMT include cisplatin and the taxanes (axonal toxicity), nitrofurantoin (long-term sensorimotor neuropathy in renal insufficiency), and metronidazole (dose-dependent sensory neuropathy with prolonged courses).
Board Trap — “A patient with CMT needs chemotherapy for leukemia: which agent is safe?”

The vignette gives you a child with known CMT1A and newly diagnosed acute lymphoblastic leukemia, and asks which induction regimen is safe. The trap is to choose any standard protocol that includes vincristine. Vincristine produces devastating, often irreversible accelerated peripheral neuropathy in CMT and must be omitted or substituted; the oncology team should be informed of the CMT diagnosis before any chemotherapy plan is finalized. Caution flags also apply to cisplatin, the taxanes, nitrofurantoin, and metronidazole. The medical alert bracelet is part of standard CMT care for exactly this reason.
CMT rehabilitation. No disease-modifying pharmacotherapy is approved for CMT, so management is rehabilitation-centric. Ankle-foot orthoses (AFOs) for foot drop are the cornerstone of mobility, with rigid plastic AFOs preferred when ankle dorsiflexion is severely weak and articulating designs preferred when modest power remains and ankle motion is desired. Daily stretching of the Achilles tendon and plantar fascia delays equinus and clawing of the toes. Strengthening is allowed but should be submaximal and concentric; eccentric overload accelerates axonal injury. Custom shoe orthotics redistribute pressure off the painful metatarsal heads created by pes cavus. Surgical correction of progressive foot deformity may include plantar fasciotomy, posterior tibial tendon transfer to the dorsum, calcaneal osteotomy for hindfoot varus, and metatarsal osteotomies. As intrinsic hand weakness emerges, hand therapy and adaptive equipment for buttons and zippers extend independence. Neuropathic pain affects 50 to 70 percent of CMT patients and responds to gabapentin, pregabalin, duloxetine, or low-dose tricyclic antidepressants.
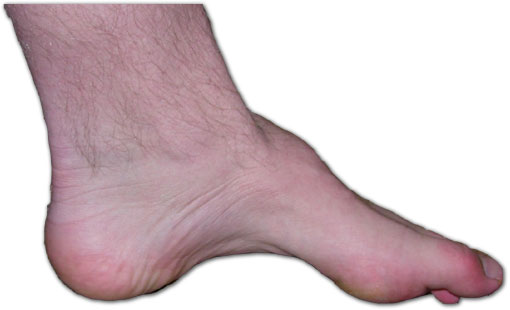
Source: Wikimedia Commons contributor, via Wikimedia Commons, CC BY-SA 3.0. https://commons.wikimedia.org/wiki/File:Charcot-marie-tooth_foot.jpg ::: {.callout-important} ## High Yield — CMT and HNPP

- CMT prevalence approximately 1 in 2,500; pes cavus, hammertoes, peroneal atrophy, steppage gait.
- Demyelinating vs axonal threshold = median motor NCV 38 m/s.
- CMT1A = most common (approximately 50 percent); PMP22 duplication 17p11.2 (gene dosage); AD; onion bulbs on biopsy.
- CMT2A = most common axonal subtype; MFN2 mutation, 1p36; AD.
- CMTX1 = X-linked, intermediate NCV; GJB1 (connexin 32).
- HNPP = reciprocal 17p11.2 deletion of PMP22; pressure-palsy phenotype; tomaculous (sausage-shaped) myelin on biopsy.
- Vincristine absolutely contraindicated in CMT (also caution with cisplatin, taxanes, nitrofurantoin, metronidazole).
- AFOs for foot drop are the rehabilitation cornerstone; surgical correction for progressive deformity.
- Neuropathic pain responds to gabapentin, pregabalin, or duloxetine. :::